The biochemical processes which life necessitates were introduced very early by evolution. The resulting living organisms are remarkably similar at the molecular level, allowing the general study of biochemistry via the observation of the uniform.
Location Hydrogen Oxygen Carbon Silicon
-------- -------- ------ ------ -------
Humans 63 25.5 9.5 < 0.1
Seawater 66 33 0.0014 < 0.1
Earth's crust 0.22 47 0.19 28
Oxygen and hydrogen are highly bioavailable as a result of the abundance of water, and their abundance in life follows. Carbon is instead far less available, but is sought out nevertheless due to its great utility.
Property Carbon Silicon
-------- ------ -------
Abundance 0.19 28
Valence electrons 4 4
Bond strength, kJ/mol 350 222
Bond selectivity Low, forms bonds freely High, prioritizes certain atoms
Bond multiplicity Single, double, triple Single, rarely double
Combustion byproduct, solubility in water Carbon dioxide, soluble Silicon dioxide, insoluble
Carbon's greater bond strength allows greater potency as a fuel. Of note is the soluble combustion byproduct of carbon, who, along with hydrogen, forms biological bonds; the gaseous state of the byproduct allows recyclability.
Biomolecules can be categorized into the four classes of proteins, nucleic acids, lipids, and carbohydrates.
Proteins are constructed from the 22 proteinogenic amino acids (of which 20 are present in the standard genetic code), which link via peptide bonds to form long, unbranched polymers. Polymers are folded into three-dimensional structures, which then facilitate biochemical functions:
Enzymes quicken the rate of reactions while expending only negligible quantities of themselves, allowing their reuse. They are a dependency of all biological systems.
Two primary themes of biochemistry are expressed via proteins: the molecular hierarchy of structure, and the dictation of function by structure.
Nucleic acids store and transfer information, primarily the instruction set for all cellular functions. They are linear molecules, like proteins. Composing nucleic acids are nucleotides, which themselves comprise a heterocyclic base ring attached both to a five-carbon sugar (either ribose or deoxyribose), and at least one phosphoryl group.
DNA (deoxyribonucleic acid), a type of nucleic acid, stores the determinant genetic information of an organism. The deoxyribonucleotide bases which comprise DNA --- adenine, cytosine, guanine, thymine --- attach to their bases via glycosidic bonds, with one, two, or three phosphoreal attached to deoxyribose. Information is stored via the seuqnece of nucleotides, linked via phosphodiesters. In all higher organisms, DNA exists structurally as double helix, in which bases interact with each other via hydrogen bonds; adenine/thymine interactions hold two hydrogen bonds, and cytosine/guanine interactions hold three.
RNA (ribonucleic acid), the other type of nucleic acid, is singly stranded, and differs significantly from DNA:
Some regions of DNA can be copied as mRNA (messenger RNA), which templates protein synthesis and is frequently decomposed after use.
Note that a nucleoside is a nucleotide deprived of phosphoreal groups.
Lipids are far smaller than proteins and nucleic acids; the latter two may possess molecular weights of thousands to millions, while a typical lipid holds a molecular weight of \(1300 \text{g mol} ^ {-1}\). Neither are lipids polymers, as proteins and nucleic acids are.
The key characteristic of many lipids is their ampipathic nature, each possessing both a hydrophilic head and a hydrophilic tail; the head is oriented toward water, while the tail is oriented away from water, and into the tails of other lipids in the case of a lipid bilayer, present in membranes. The hydrophobic tail of lipids can also combust, allowing lipids to serve as fuels.
Note that lipids are also crucial signal molecules.
Carbohydrates are a principal biological fuel source, the mots common form of which is glucose. In animals, glucose composes glycogen, a branching tree of glucose molecules; plants store glucose as starch, which is similar in composition to glycogen.
Thousands of unique carbohydrates exist, which can be linked in chains and branched even moreso than in glycogen or starch. Many components of the cell exterior are decorated with carbohydrates, which can be identified by other cells; notably, the arrangement of carbohydrates on red blood cells dictates one's blood type.
Cellular information processing complexity compounds as cells form tissues and tissues form organisms. In 1958, Francis Crick first proposed the underlying scheme of gene expression, referred to as the central dogma of biology: DNA replicates itself; DNA forms RNA via transcription; RNA forms proteins via translation. Despite the existence of exceptions, the core tenets of the central dogma hold.
DNA constitutes the heritable information of an organism (the genome) packaged into discrete units (genes). During cell duplication, DNA is copied to place identical genomes into the newly formed daughter cells. DNA polymerase, an enzyme collective, catalyzes replication.
In a similar fashion, RNA polymerase catalyzes the transcription from DNA to RNA, allowing gene expression. The choice of genes transcribed, and the spatial and temporal details of transcription, are crucial to the fate of the cell. Each cell in the human body has the information to form any tissue; transcription allows only the necessary details to be expressed.
Translation, occurring on ribosomes (which themselves consist of RNA and proteins) renders functional the genetic information within mRNA, in the form of proteins.
Note that, contrary to the central dogma, reverse transcriptase allows the reversal of transcription.
Organisms vary in complexity, from one cell to trillions; each cell is delineated by a membrane comprising a lipid bilayer.
A eukaryotic cell, the first type of basic cell, encloses organelles in membrane compartments. A prokaryotic cell, the other type of basic cell, is surrounded by two membranes, separated themselves by a periplasmic space. It lacks compartmentalization, and is quite simple in comparison to a eukaryotic cell.
Humans comprise roughly \(40\) trillion eukaryotic cells, and we each carry an equivalent number of prokaryotes, collectively our microbiome. Intestinal prokaryotes assist in digestion, provide nutrition (including vitamins), and defend against less benevolent microorganisms. Disruption of the microbiome is linked to development of type 2 diabetes, liver disease, cardiovascular disease, and inflammatory bowel disease.
Two features minimally constitute a cell, regardless of other qualities: there must be a barrier which separates the cell from its environment; and the interior of the cell must be chemically different from its environment, and accomodate the biochemistry of the organism. This barrier is the plasma membrane, and the fundamental intracellular material is the cytoplasm.
The plasma membrane separates the interior of the cell from its environment. It is impermeable to most substances, even those critical to the cell's function (such as fuels, building blocks, and signal molecules); its semipermeability must be regulated to selectively allow transmission of molecules to and from the cell. The regulatory proteins which facilitate this selectivity allow the entry of fuels (such as glucose) and building blocks (such as amino acids); they additionally transduce information (such as insulin).
The plasma membrane of a plant cell is bulwarked by a cell wall, comprising mostly a linear glucose polymer known as cellulose, molecules of which interact with one another as well as the other components of the cell wall to protect the cell.
The cytoplasm comprises the inner substance of a cell. It hosts many biochemical processes; some are the initial stage of glucose metabolism, fatty acid synthesis, and protein synthesis. The cytoplasm is carefully organized by the cytoskeleton, a network of structural filaments. The cells of many eukaryotes have the three types of protein fibers --- actin filaments, intermediate filaments, and microtubules --- which support the structure of the cell, localize biochemical activities, and enable intracellular molecular transport.
It is only eukaryotes who possess organelles, a complex array of intracellular, membrane-bound compartments.
The nucleus, bound by a double membrane, is the largest organelle. As it holds the genome of the organism, it can be called the information center of the cell. The nuclear membrane is punched with pores which permit transport to and from the nucleus, facilitating the movement of DNA and RNA, which are synthesized in the cytoplasm but function in the nucleus. Genomic information is selectively expressed in the nucleus.
The mitrochondrion, like the nucleus, is bound by two membranes --- an outer membrane in contact with the cytoplasm, and an inner membrane which defines the mitrochondrial matrix --- the space between which is the intermembrane space. Mitrochondria combusts fuel molecules to generate ATP. Roughly \(90\%\) of a cell's energy is produced by the mitochondrion; poisons such as cyanide and carbon monoxide are toxic due to their inhibition of the mitrochondrion.
Found only in plant cells, the chloroplast is also bound with a double membrane. The chloroplast, being the site of photosynthesis, is essential to all life; if photosynthesis were to halt, life on Earth would cease in roughly \(25\) years.
The ER (endoplasmic reticulum) is a series of membranous sacs (the interior of which are lumens), upon or within which a great number of biochemical reactions take place.
The smooth ER, the first type of ER, is most notable for its processing of exogenous chemicals, such as drugs. THe quantity of drugs ingested by an organism correlates to the number of smooth ER in its liver.
The rough ER, the other type of ER, is named for the ribosomes which populate its exterior. Rather than synthesizing proteins for use within the cell, ribosomes attached to the rough ER synthesize proteins for use either outside the cell or in the cell membrane. Proteins synthesized on the rough ER are transported into the lumen during translation, within which a protein is folded into its final three-dimensional structure via the assistance of chaperone proteins, and modified via the attachment of carbohydrates. These modified proteins are then sequestered into ribosome-deprived regions of the rough ER, which bud off of the rough ER as transport vesicles.
Rough ER transport vesicles are carried to the Golgi complex, a series of stacked membranes, and fuse with it for further processing via the addition of a different set of carbohydrates, and for sorting by destination.
When a vesicle filled with proteins destined for secretion buds off of the Golgi complex, a secretory granule is formed and directed toward the cell membrane. Upon the proper signal, the granule fuses with the membrane and expels its contents into the environment via exocytosis.
Endocytosis, the subsummation of material by the cell, occurs when the plasma membrane invaginates and buds off to form an endosome. The process is critical in the collection of biochemicals such as iron, cobalamin, and cholestrol into the cell. Endocytosis occurs through small regions of the membrane when a small amount of material must be taken in, or through a modified process called phagocytosis for a greater amount of material.
Lysosomes contain a wide array of digestive enzymes. They form analogously to secretory granules, forming with endosomes rather than the plasma membrane, and digest material after fusion has taken place, releasing small molecules for use by the cell. Digestion of damaged organelles is also a role of the lysosome.
Also unique to plant cells, the vacuole occupies up to \(80\%\) of the volume of a cell, and stores nutrients, water, and ions. Proteins transport molecules across the vacuolar membrane.
The complex structures and functions of cells are stabilized by weak interactions with only a fraction the strength of covalent bonds. The possibility of stabilization by such weak interactions is due to the great number of these interactions taking place; and the functions of life necessitate these weak bonds to allow transient interactions: a substrate can bind to an enzyme, and the product may leave unhindered.
Water affects weak bonds to a high degree, either weakening or strengthening them. The inability of water to interact with some molecules at all is also crucial, allowing the folding of proteins and the compartmentalization of cells.
The human experience of life occurs at a distance of \(0.4 \text{ nm}\) (\(4 \text{\r{A}}\)), the typical length of weak interactions.
Brownian motion, discovered by Robert Brown, is a source of particle movement as a result of the random fluctuation of thermal noise. As Brownian motion does not expend energy, it is responsible for initiating many biochemical interactions. In the cell, water is the most common medium for Brownian motion, facilitating energy and information transfer and enabling enzymes to find substrates, modifications of fuels, and diffusions of signal molecules.
Cells are complex; organization of the cell is necessary to facilitate useful Brownian motion.
Water, the solvent of life, composes \(65\%\) of each human being and about \(70\%\) of each cell. In liquid water, hydrogen bonds constantly break and reform; the average liquid water molecule has hydrogen-bonded to \(3.4\) neighboring water molecules. This grants water the cohesion to perform feats such as the transport of material up tall trees via transpiration; the height of trees is likely limited by the strength of hydrogen bonds between water molecules, as the breakage of these bonds creates a bubble, or embolism, which prevents further water flow.
Crucially, water is able to dissolve essentially any polar solute, and similarly important is the inability to dissolve nonpolar compounds. The hydrophobic effect, spontaneously occurring due to entropy, causes the layered separation of water and nonpolar material, which is used extensively for protein folding and the formation of membranes.
Noncovalent, reversible molecular interactions are crucial in the flow of energy and information, enabling accurate DNA replication, the folding of proteins, reactant recognition by enzymes, and molecular signal detection.
The three fundamendal noncovalent bonds provide for transient biochemical interactions: ionic bonds (electrostatic interactions), hydrogen bonds, and van der Waals interactions.
Also called ionic bonds or salt bridges electrostatic interactions comprise the interactions between the distinct electrical charges on atoms. The energy of an electrostatic interaction between two ions is given by Coloumb's law, \(E=\frac{k q_1 q_2}{Dr}\), where:
The electrostatic interaction betwween two atoms bearing single, opposite charges varies inversely with both the square of the distance between them and the nature of the intervening medium.
Property Water Hexane Vacuum
-------- ----- ------ ------
Dielectric constant 80 2 1
Electrostatic interaction energy in kJ/mol -5.8 -231 0
Electrostatic interactions are therefore strongest in a vacuum, in which the dielectric constant is lowest; water weakens electrostatic interactions by outcompeting ionic bonds, bonding directly to the ions. Water can, in this way, dissolve essentially any molecule with charge great enough to interact with. Along this line, the strength of electrostatic interactions is maximized in an uncharged environment.
Ionic bonds maintain a bond length of roughly \(3 \text{\r{A}}\).
Hydrogen bonds can arise whenever hydrogen is covalently bound to an electronegative atom, most often nitrogen or oxygen.
Property Covalent Hydrogen
-------- -------- --------
Bond energy, kJ/mol about 418 8 to 20
Hydrogen bonds, measured from the hydrogen atom, measure from \(1.5\) to \(2.6\) \(\text{\r{A}}\); distances ranging from \(2.4\) to \(3.5\) \(\text{\r{A}}\) therefore separate the two nonhydrogen atoms in a hydrogen bond. In aqueous solution, hydrogen bonds will be disrupted by water via outcompetition.
A great host of essential biomolecules are neither polar nor charged; interaction amongst them is permitted by van der Waals interactions.
The distribution of electronic charge around an atom varies randomly over time as charge distribution is not perfectly symmetric at any given instant; random regions of partial charges thus arise, which can fuel electrostatic interactions to induce complementary asymmetry in toe electron distribution around its neighboring atoms. Affected atoms continue toward one another until they reach the van der Waals contact distance (about \(3\) to \(4\) \(\text{\r{A}}\)), at which point repulsive forces dominate attractive forces as electron clouds begin to overlap.
van der Waals interaction energies typically contribute \(2\) to \(4\) \(\text{kJ mol} ^ {-1}\). While quite weak, they are most significant when involving large molecules with complementary shapes, allowing a great number of atoms in contact. Using the van der Waals between their feet and the surface, geckos are able to climb up walls and across ceilings.
The transient nature of weak bonds is crucial to biochemical processes. As an example, hydrogen bonds between the base pairs of DNA stabilize the double helix structure, keeping the base sequence protected from destructive reactions by the helix structure; the weak nature of hydrogen bonds permit the helix to be opened to replicate or express DNA.
Nonpolar molecules are unable to interact with water, instead forming isolated cavities, and the ordering of water around these clusters reduces entropy. Minimal surface area between water and nonpolar material, leading to the least possible ordered water molecules, is therefore the outcome of nonpolar/water interactions, as the increase of entropy is a spontaneous process. A consequence of this, of great importance to biochemistry, is the absence of an energy cost to the use of the hydrophobic effect.
The ampipathic (both hydrophilic and hydrophobic) nature of membrane phospholipids allows the automatic orientation of the head toward, and the tail away from, the aqueous medium. The tails of the lipid bilayer, caged by the heads, interact with one another, forming a membrane. Membranes are further stabilized by van der Waals forces between the hydrophobic tails of each layer.
Proteins are permitted to play prominent roles in all aspects of energy and information manipulation by their capability to form complex, three-dimensional structures which allow specific interactions with other biomolecules.
The folding of proteins drives a loss of entropy which is counterbalanced by the hydrophobic effect. The nonpolar groups present in proteins strongly tend to associate with one another in the interior of the folded protein; the resulting increase of entropy in water compensates for the entropy losses of the folding process.
The hydrophobic effect powers the folding of proteins, but its stabilizing properties are replaced with weak attractions (hydrogen bonds and van der Waals interactions) in the unfolded protein.
Generalizations exist which make the analysis of the sheer quantity of biomolecules easier. We can classify biomolecules into proteins, nucleic acids, lipids, and carbohydrates; and we can further identify characterizing functional groups with which to classify biomolecules:
Name Class(es) Formula(s)
---- --------- ----------
Hydrophobic Alipathic, Aromatic R-Me, R-Ph
Hydroxyl Alcohol R-OH
Aldehyde Aldehyde R-CHO
Keto Ketone R-CRO
Carboxyl Carboxylic acid R-COOH
Amino Amine R-NH_2
Phosphate Organic phosphate R-OPO_3
Sulfhydryl Thiol R-SH
Reversible acid-base reactions, those which include the release or binding of a proton, are biochemically crucial. The \(\text{pH}\) of a solution is the measure of its proton concentration, and ranges from \(0\) to \(14\), with small numbers expressing an acidic environment, and large numbers a basic environment.
Maintaining a certain \(\text{pH}\) is of great biological importance: in humans, deviations of \(\pm 0.5 \text{ pH}\) from the standard \(\text{pH}\) of blood, \(7.4\), can lead to coma and death. Improper alterations of \(\text{pH}\) have a great effect on the electrostatic environment of an organism, which can disrupt crucial weak interactions. Biomolecules affected in this way often lose their function as a result of the alteration of their structure.
Small amounts of pure water dissociate to form hydronium (\(\text{H}_3\text{O}^+\), also represented as \(\text{H}^+\)) and hydroxyl (\(\text{OH}^-\)) ions, the concentration of each being \(10^{-7} \text{ M}\).
\[\text{H}_2\text{O} \rightleftharpoons \text{H}^+ + \text{OH}^-\]
The equilibrium constant, \(K_{eq}\), of the above is given by:
\[K_{eq} = \frac{[\text{H}^+][\text{OH}^-]}{\text{H}_2\text{O}}\]
The brackets above denote molecular concentrations, \(\text{M}\), of the molecules. Substituting concentrations for \(\text{H}^+\), \(\text{OH}^-\) (\(10^{-7} \text{ M}\) each), and \(\text{H}_2\text{O}\) (\(55.5 \text{ M}\)) gives:
\[K_{eq} = \frac{\left(10^{-7} \text{ M}\right)^2}{55.5 \text{ M}} = 1.8 \times 10^{-16} \text{ M}\]
The concentration of water is essentially unchanged by the small degree of ionization; we can therefore ignore changes in the concentration of water to define a new constant, \(K_w\), the ion product of water:
\[K_w = K_{eq} \times [\text{H}_2\text{O}] = [\text{H}^+][\text{OH}^-]\] \[T = 25^\circ \text{ C} \implies K_w = 1.0 \times 10^{-14} \text{ M}^2\]
The \(\text{pH}\) and \(\text{pOH}\) of a solution are defined as:
\[\text{pH} = -\log_{10}([\text{H}^+])\] \[\text{pH} = -\log_{10}([\text{OH}^-])\]
Pure water, which contains an equal mass each of \(\text{H}^+\) and \(\text{OH}^-\), has a \(\text{pH}\) (and \(\text{pOH}\)) of \(7\). As the concentrations of \(\text{H}^+\) and \(\text{OH}^-\) are reciprocally related, \(\text{pH} + \text{pOH} = 14\) for any solution.
Acids, incredibly prominent in biochemistry, ionize to produce a proton and a base. Formed by the ionization of an acid is its conjugate base; in the same fashion, the protonation of a base produces its conjugate acid.
\[\overbrace{\text{CH}_3\text{COOH}}^{\text{Acetic acid}} \rightleftharpoons \text{H}^+ + \overbrace{\text{CH}_3\text{COO}^-}^{\text{Acetate}}\]
The ionization equilibrium of a weak acid, \(\text{HA}\), is given by:
\[\text{HA} \rightleftharpoons \text{H}^+ + \text{A}^-\]
The equilibrium constant for the above ionization, \(K_a\), is given by:
\[K_a = \frac{[\text{H}^-][\text{A}^-]}{[\text{HA}]}\]
We can describe the relationship between \(\text{pH}\) and the acid-to-base ratio of a solution:
\[\frac{1}{[\text{H}^+]} = \frac{1}{K_a}\frac{[\text{A}^-]}{[\text{HA}]} \implies \log_{10}\left(\frac{1}{[\text{H}^+]}\right) = \log_{10}\left(\frac{1}{K_a}\right)\log_{10}\left(\frac{[\text{A}^-]}{[\text{HA}]}\right)\]
We define the \(\text{p}K_a\) of the acid to be \(\log_{10}\left(\frac{1}{K_a}\right)\).
The Henderson-Hasselbalch equation relates \(\text{pH}\) and \(\text{p}K_a\):
\[\text{pH} = \text{p}K_a + \log\left(\frac{[\text{A}^-]}{[\text{HA}]}\right)\]
An acid's \(\text{p}K_a\) is the \(\text{pH}\) at which it dissociates halfway. Above an acid's \(\text{p}K_a\), \([\text{A}^-]\) dominates the equilibrium; below it, \([\text{HA}]\) dominates. Most crucial biochemical acids are dissociated at physiological \(\text{pH}\) (roughly \(7.4\)), and these molecules are therefore typically referred to as their conjugate base.
Conjugate acid-base pairs act as buffers by resisting changes to the \(\text{pH}\) of a solution. Take the addition of \(\text{OH}^-\) to a solution of acetic acid (\(\text{HA}\)):
\[\text{HA} + \text{OH}^- \rightleftharpoons \text{A}^- + \text{H}_2\text{O}\]
The plot of \(\text{pH}\) against the added amount of \(\text{OH}^-\), the titration curve, indicates an inflection point at \(\text{pH } 4.8\), the \(\text{p}K_a\) of acetic acid. In the same fashion, a weak acid is most effective in buffering against \(\text{pH}\) in the vicinity of its \(\text{p}K_a\).
Biochemical processes require buffers to regulate \(\text{pH}\) and therefore maintain regular function. In respiration, for example, carbon dioxide is produced, reacting with water to form carbonic acid, in turn dissociating into a proton and a bicarbonate ion:
\[\text{CO}_2 + \text{H}_2\text{O} \rightleftharpoons \text{H}_2\text{CO}_3 \rightleftharpoons \text{H}^+ + \text{HCO}_3^-\]
Here, the conjugate acid-base pair of \(\text{H}_2\text{CO}_3\) and \(\text{HCO}_3^-\) acts as a buffer; protons released by an external acid will be captured by the bicarbonate ion, preventing significant alteration of \(\text{pH}\). This maintenance of the \(\text{pH}\) of blood is coined compensatory respiratory alkalosis.
Notably, the equilibrium between \(\text{CO}_2\) and \(\text{H}_2\text{CO}_3\) is incredibly rapid; \(\text{CO}_2\) is often referred to as the conjugate acid of \(\text{HCO}_3^-\).
Buffer problems can be solved using the Henderson-Hasselbalch equation:
\[\text{pH} = \text{p}K_a + \log\left(\frac{[\text{A}^-]}{[\text{HA}]}\right)\]
We want to examine how to make \(1 \text{ L}\) of \(0.3 \text{ M}\) acetate buffer with \(\text{pH } 4.46\), with, on hand, \(2 \text{ M}\) acetic acid (\(\text{p}K_a = 4.76\)) and \(2.5 \text{ M}\) potassium hydroxide (\(\text{KOH}\), a strong base). We must first determine the ratio of acetate to acetic acid which will yield \(\text{pH } 4.46\), then we can calculate the amount of each reactant needed to yield a molarity of \(0.3 \text{ M}\).
\[4.46 = 4.76 + \log\left(\frac{[\text{acetate}]}{[\text{acetic acid}]}\right)\] \[-0.3 = \log\left(\frac{[\text{acetate}]}{[\text{acetic acid}]}\right)\] \[\frac{[\text{acetate}]}{[\text{acetic acid}]} = 0.5\]
We can therefore conclude that, to obtain \(\text{pH } 4.46\) with the acetate buffer, \(2/3\) (\(0.2 \text{ M}\)) of the acetate will be contributed by the acid, and \(1/3\) (\(0.1 \text{ M}\)) by the base.
Amino acids serve a great host of biochemical processes; among them are their function as signal molecules (such as neurotransmitters) and their prototyping to other biomolecules (such as hormones, nucleic acids, lipids, and proteins).
A projection is required to depict a three-dimensional molecule on a two-dimensional page; two are used to depict amino acids.
In a Fischer projection, each atom is identified, and the bonds to the central carbon atom are represented by horizontal and vertical lines. Horizontal bonds indicate projection out of the page (toward the viewer), and vertical bonds projection into the page (away from the viewer).
In a stereochemical rendering, carbon and hydrogen are not indicated unless crucial to the function of the molecule. Straight lines depict bonds parallel with the plane of the page, with a wedged bond indicating projection out of the page, and a dashed bond projection into the page.
Amino acids compose proteins. An \(\alpha\)-amino acid comprises a central \(\alpha\)-carbon bonded to an amino group, a carboxylic acid group, a hydrogen atom, and a unique side chain, called the \(\text{R}\) group.
\(\alpha\)-amino acids are chiral; they may exist in either of two mirrored forms, called the \(\text{L}\) and \(\text{D}\) isomers. But, only \(\text{L}\) isomers compose proteins. The tailoring of biomolecules to interact with only one isomer proved more efficient; it is likely the \(\text{L}\) isomer was chosen by chance.
Free amino acids in solution, at neutral \(\text{pH}\), are zwitterionic: the amino group is protonated (\(\text{NH}_3^+\)), and the carboxyl group is deprotonated (\(\text{COO}^-\)). The ionization state of an amino acid varies: at \(\text{pH} \approx 1\), the amino group is protonated (\(\text{NH}_3^+\)), and the carboxyl group is not dissociated (\(\text{COOH}\)); as \(\text{pH}\) is raised, the carboxylic acid is deprotonated first at \(\text{pH} \approx 2\), then the deprotonation of \(\text{NH}_3^+\) follows at \(\text{pH} \approx 9\).
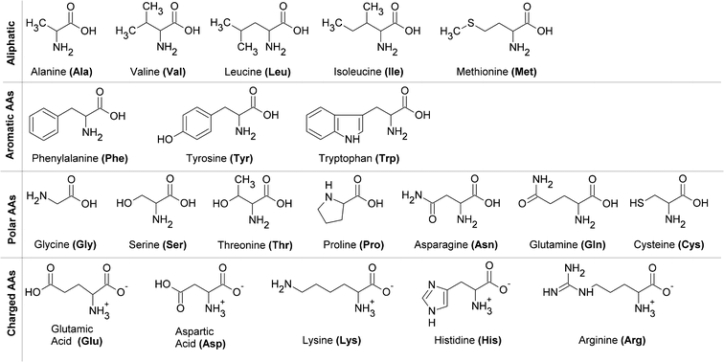
All proteins of all species are constructed from the same set of \(20\) proteinogenic amino acids, which vary only in their \(\text{R}\) groups, each containing any of an array of functional groups. We may sort these amino acids by the chemical characteristics of their \(\text{R}\) groups:
Glycine is, notably, the only achiral amino acid, with a lone hydrogen atom as its side chain. Proline is also unique in that its side chain attaches both to the nitrogen and the \(\alpha\)-carbon of its backbone, conformationally restricting the proteins it composes.
Two of the hydrophobic amino acids, phenylalanine and tryptophan, have aromatic side chains: phenylalanine's \(\text{R}\) group is a phenyl ring, and tryptophan's is an indole ring joined to a methylene group.
The hydrophobic amino acids tend to pack together within the protein as a result of the hydrophobic effect, with the great host of hydrophobic amino acids permitting more efficient clustering due to varied conformations.
The hydroxyl groups of serine, threonine, and tyrosine offer great hydrophilicity, which permits interaction with water in the protein-folding process.
Cysteine contains the highly reactive sulfhydryl group, which quickly ionizes at slightly basic \(\text{pH}\) to form a thiolate. Pairs of sulfhydryl groups may form disulfide bonds, which prove crucial in the stabilization of some proteins.
The \(\text{R}\) groups of lysine and arginine have dual properties, as their carbon chains constitute a backbone, but each chain is terminated with a positive charge. Histidine contains an aromatic imidazole group, which uniquely may either be neutral or positive near neutral \(\text{pH}\), offering utility in catalysis.
Aspartate and glutamate each may accept protons, which neutralizes their charge. They are closely related to asparagine and glutamine, respectively, in which a carboxylic acid in the former pair replaces a carboxamide in the latter.
Seven of the twenty proteinogenic amino acids have ionizable side chains:
The varied \(\text{p}K_a\) of these groups allows similarly varied roles in acid-base catalysis.
Humans can only manufacture eleven of the proteinogenic amino acids, with the remaining nine being obtained via diet. Amino acid production also depends on factors such as age; an adult can synthesize arginine as needed, while a child requires additional arginine from the environment.
Proteins are the transition from the one-dimensional view of microbiomolecules to the three-dimensional view of complex biological systems. DNA encodes the sequence of fifty to two thousand amino acids (the primary structure) which constitutes a protein.
Proteins are not linear; they fold via the hydrophobic effect to form three-dimensional structures which dictate their respective functions
The secondary structure of proteins arises from a regular pattern of hydrogen bonds between the \(\text{NH}\) and \(\text{CO}\) groups which comprise the amino acids in the polypeptide chain.
Tertiary structure, the highest level of structure an individual polypeptide can obtain, becomes apparent as the \(\text{R}\) groups of amino acids with great distance between them bond to one another.
Quaternary structure may be achieved by proteins which require multiple chains to function; it may be as simple as a dimer of two identical polypeptides or as complex as a polymer of dozens of unique polypeptides.
The three-dimensional structure of proteins relies solely on the primary structure: the polymers formed by linking the \(\alpha\)-carboxyl of one amino acid to the \(\alpha\)-amino of another via dehydrative peptide (or amide) bonds. The peptide bond reaction favors hydrolysis rather than synthesis, and formation of dipeptides therefore requires free energy. Peptide bonds, regardless, are highly stable kinetically; their lifetimes approach one thousand years in the absence of catalysis.
Each amino acid unit in a polypeptide is called a residue. Polypeptide chains have directionality (or polarity), beginning at the \(\alpha\)-amino and ending at the \(\alpha\)-carboxyl. Amino acids are therefore written beginning with the amino-terminal residue; in the pentapeptide \(\text{YGGFL}\), tyrosine is the amino-terminal (or \(\text{N}\)-terminal), and leucine is the carboxyl-terminal (or \(\text{C}\)-terminal).
A polypeptide chain consists of a backbone, shared among polypeptides, and the side chains of each amino acid, which is unique. The backbone, with a carbonyl and amino group per residue, hosts great hydrogen-bonding capability.
Most polypeptide chains, referred to as proteins, comprise \(50\) to \(2000\) amino acids. The largest known protein, titin, consists of nearly \(27000\) amino acids, and scaffolds the assembly of muscle proteins.
The mean molecular weight of amino acid residues is roughly \(110 \text{ g mol}^{-1}\); the molecular weights of most proteins are therefore between \(5500\) and \(22000 \text{ g mol}^{-1}\). Note that the dalton (\(\text{Da}\)) represents a mass equivalent to \(1 \text{ g mol}^{-1}\): a protein with molecular weight of \(50000 \text{ g mol}^{-1}\) has a mass of \(50 \text{ kDa}\).
The linear polypeptide is sometimes cross-linked, frequently via the disulfide bonds formed by the oxidation of a pair of cysteine residues, the resulting unit of which is coined cystine. Disulfide bonds may link together cysteines of a single polypeptide, or they may connect two separate chains together.
The amino acid sequence of insulin was discovered by Fredireck Sanger in 1953: it showed for the first time that each protein has a definite amino acid sequence, consisting only of \(\text{L}\) amino acids linked via peptide bonds. A landmark in biochemistry, it paved the way for the millions of protein sequences which were discovered since.
It is primarily amino acid sequences which determine the three-dimensional structure of their respective proteins: knowledge of the sequence of a protein is essential to elucidating its function. Alterations in primary structure can lead to insufficient function, then disease; learning the sequence can allow development of treatments. The sequence of a protein can also reveal much of its evolutionary history, permitting the budding field of molecular paleontology to flourish.
A great host of factors determine the relation between the primary and tertiary structure of a protein. The peptide bond is essentially planar: a dipeptide places six atoms (the \(\alpha\)-carbon and \(\text{CO}\) of the first amino acid, and the \(\text{NH}\) and \(\alpha\)-carbon of the second) in the same plane.
The peptide bond also has considerable double bond character due to the resonance of the amide, preventing rotation around the bond and therefore constrains the conformation of the peptide backbone. This character is expressed in the length of the bond between \(\text{CO}\) and \(\text{NH}\): the \(\text{C} - \text{N}\) peptide bond distance is roughly \(1.32 \text{ \r{A}}\), which is between the typical bond lengths for a \(\text{C} - \text{N}\) bond (\(1.45 \text{ \r{A}}\)) and a \(\text{C} = \text{N}\) bond (\(1.27 \text{ \r{A}}\)). The peptide bond is uncharged, which permits polypeptides to form packed structures which would otherwise be prohibited by charge repulsion.
A planar peptide bond has two possible conformations: the trans configuration pushes the two \(\alpha\)-carbons to opposite sides of the bond, while the cis configuration instead places the \(\alpha\)-carbons on the same side of the bond. Steric clashes often prohibit the cis configuration; nearly every protein peptide bond is trans.
The amino and carbonyl of an amino acid are singly bonded to the \(\alpha\)-carbon, permitting the rotation of adjacent, rigid peptide units around these bonds. This freedom of rotation offers great flexiblity to protein folding. The rotations around these bonds can be specified by torsion angles: the angle of rotation between the nitrogen and the \(\alpha\)-carbon atom is referred to as \(\phi\), while the angle of rotation between the \(\alpha\)-carbon and the carbonyl is coined \(\psi\). A clockwise rotation about either bond, as viewed towards the \(\alpha\)-carbon, corresponds to a positive value. The angles \(\phi\) and \(\psi\) determine the path of the polypeptide.
Not all combinations of \(\phi\) and \(\psi\) are possible: the Ramachandran plot (named for Gopalasamudram Ramachandran) illustrates the \(\phi\) and \(\psi\) values of possible conformations.
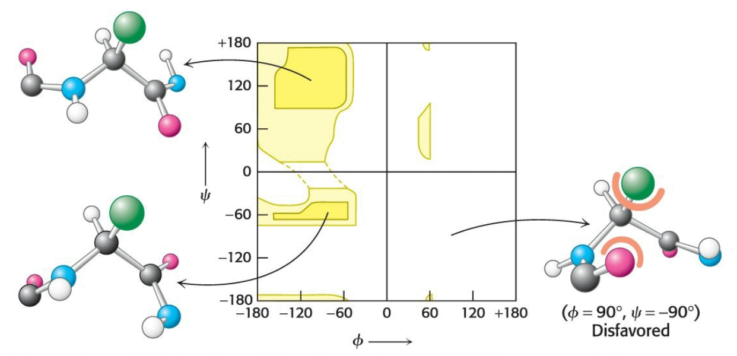
Steric exclusion acts as a powerful organizer via the restriction of the number of possible peptide conformatons.
In 1951, Linus Pauling and Robert Corey proposed that some polypeptides have the ability to fold into two periodic structures: the \(\alpha\) helix and the \(\beta\) pleated sheet. Other structures, such as turns and loops, have been identified since; each is formed by a regular pattern of hydrogen bonds between the aminos and carboxyls which appear in close proximity to on eanother in the primary structure.
The side chains of the amino acids which compose an \(\alpha\) helix extend outward in a helical array, forming a tightly coiled backbone. The helix is stabilized via hydrogen bonds between the amino and carboxyl groups of the chain: the carboxyl of each amino acid hydrogen-bonds to the amino group of the amino acid situated four residues ahead in the linear sequence. Singular \(\alpha\) helices are usually less than \(45 \text{ \r{A}}\) long.
Each residue is linked to the next via a rise (or translation) of \(1.5 \text{ \r{A}}\) and a rotation of \(100^\circ\), giving \(3.6\) residues per helix turn: residues spaced three to four apart in the linear sequence are in closer proximity to one another than those spaced two apart. The helix pitch is the length of one turn around the helix axis, and is expressed by the product of the translation (\(1.5 \text{ \r{A}}\)) and the number of residues per turn (\(3.6\)), giving \(5.4 \text{ \r{A}}\).
Helices can turn clockwise (right-handed) or counterclockwise (left-handed). Right-handed helices are energetically favorted due to fewer steric clashes between the side chains and the backbone: essentially all \(\alpha\) helices found in proteins are right-handed.
Amino acids which branch at the \(\beta\)-carbon tend to destabilize \(\alpha\) helices due to steric clashes: valine, threonine, and isoleucine. Serine, aspartate, and asparagine tend to disrupt \(\alpha\) helices due to their outcompetition of the main chain for hydrogen bonds. Proline is often absent from \(\alpha\) helices due to the great conformational restriction it presents.
The \(\alpha\)-helical content or proteins ranges widely; for example, roughly \(75\%\) of the residues in ferritin are found in \(\alpha\) helices. About \(25\%\) of all soluble proteins are compososed of \(\alpha\) helices.
The \(\beta\) pleated sheet (or \(\beta\) sheet), rather than a single polypeptide strand, comprises multiple polypeptide chains termed \(\beta\) strands. Unlike \(\alpha\) helices, \(\beta\) strands are almost completely extended: the distance between adjacent residues along a \(\beta\) strand is approximately \(3.5 \text{ \r{A}}\), as opposed to the distances of \(1.5 \text{ \r{A}}\) along an \(\alpha\) helix. The side chains of adjacent residues point in opposite directions.
A \(\beta\) sheet is formed via the hydrogen-bonding of multiple adjacent \(\beta\) strands. Adjacent chains of a \(\beta\) can run in either identical (parallel) or opposite (antiparallel) directions. \(\beta\) sheets typically comprise \(4\) to \(5\) strands, but can instead comprise up to \(10\); these sheets can be entirely parallel, entirely parallel, or a mixture of the two.
Unlike \(\alpha\) helices, \(\beta\) sheets can consist of sections of a polypeptide which are not in close proximity to one another; in two \(\beta\) strands which lie adjacent, the final residue of the former strand and the first residue of the latter strand need not be neighbors in the linear sequence.
\(\beta\) sheets are usually twisted. In three-dimensional representations, \(\beta\) strands are typically depicted via arrows pointing to the \(\text{C}\)-terminus to indicate the parallelism of the composed sheet.
The \(\beta\) sheet is a crucial structural element; for example, fatty-acid-binding proteins are composed nearly only from \(\beta\) sheets.
Most proteins adopt compact, globular shapes, which require reversals of the orientations of their respective polypeptide chains. Reverse turns and loops permit these reversals, and these structural elements lie invariably on the surface of proteins. Loops exposted to an aqueous environment typically comprise hydrophilic residues.
Fibrous proteins serve a structural role via the formaton of long fibers, the three-dimensional structure of which is typically simple.
\(\alpha\)-Keratin, the primary component of wool and hair, comprises two right-handed \(\alpha\) helices which intertwine to form a left-handed superhelix named a coiled coil. \(\alpha\)-Keratin is a member of a protein superfamily designated the coiled-coil proteins, within each of which multiple helices entwine to form a highly stable structure with a length which exceeds \(1000 \text{ \r{A}}\). Humans have about \(60\) members of this superfamily, including intermediate filaments (of cytosol) and the muscle proteins myosin and tropomyosin. The two helices in \(\alpha\)-keratin are cross-linked via weak interactions and disulfide bonds.
A different type of helix is present in collagen, the most abundant mammalian protein and the primary fibrous component of skin, bone, tendon, cartilage, and teeth. It contains three helical polypeptides, each nearly \(1000\) residues long: glycine appears at every third residue in the linear seuqence, and the sequence glyine-proline-proline recurs frequently.
The hydrogen bonds within each peptide chain are absent in this type of helix; instead, helices are stabilized by steric repulsion of the pyrrolidine rings of the proline residues. These rings avoid each other as the polypeptide adopts its helical form, with about \(3\) residues per turn. \(3\) strands wind around one another to form a superhelical cable which is stabilized via hydrogen bonds between the \(\text{NH}\) groups of glycine residues and the \(\text{CO}\) groups of other residues. The interior of the triple-stranded helical cable is very crowded: the only residue that can fit into an interior position is glycine. Either residue which flanks glycine exists on the exterior of the cable.
Tertiary structure refers to the spatial arrangement of far-apart residues and of the pattern of disulfide bonds, resultant of the interactions between the \(\text{R}\) groups of the peptide chain.
In contrast with fibrous proteins such as keratin, globular proteins such as myoglobin have a compact three-dimensional structure and are water-soluble. Globular proteins, with their more intricate three-dimensional structure, perform most chemical transactions in the cell.
Myoglobin, a single polypeptide chain of \(153\) amino acids, is an oxygen-binding protein found primarily in heart and skeletal muscle which facilitates the diffusion of oxygen from the blood to the mitochondria, the primary site of oxygen utilization in the cell. The capacity of myyoglobin to bind oxygen correlates to the presence of heme, a prosthetic group centered by an iron atom.
Myoglobin is an extremely compact molecule: its overall dimensions are \(45 \times 35 \times 25 \text{ \r{A}}\), an order of magnitude less than if it were fully stretched out. About \(70\%\) of the main chain is folded into eight \(\alpha\) helices, and much of the rest of the chain forms turns and loops between helices.
Myoglobin, like most other proteins, is asymmetric due to the complex folding of its main chain. A unifying principle emerges from the distribution of side chains: the interior consists almost entirely of nonpolar residues. The only polar residues within the protein are two histidine residues, which play critical roles in binding the heme iron to oxygen. The exterior of myoglobin comprises both nonpolar and polar residues, rendering the molecule water-soluble. Very little empty space exists within myoglobin.
In an aqueous environment such as the interior of the cell, protein folding is driven by the hydrophobic effect: the polypeptide folds such that its hydrophobic side chains are buried and its polar chains are exposed. An unpaired \(\text{NH}\) or \(\text{CO}\) of a residue of the main chain prefer water to a nonpolar environment; the only way to bury part of the main chain within a hydrophobic atmosphere is to pair all \(\text{NH}\) and \(\text{CO}\) groups via hydrogen bonds. This pairing is accomplished in an \(\alpha\) helix or a \(\beta\) sheet.
Some proteins which span biological membranes are considered the exceptions which prove the rule: they have the reverse distribution of hydrophobic and hydrophilic amino acids. Porins, which appear on the outer membranes of many bacteria, are covered by hydrophobic residues which interact with the hydrophobic environment; in contrast, the center of the protein contains many charged, polar amino acids which surround a water-filled channel which runs through the middle of the protein. As porins function in hydrophobic environments, their composition is inside out relative to proteins which function in aqueous solution.
Certain combinations of secondary structure, called motifs or supersecondary structures, are present in many proteins and frequently exhibit similar functions; for example, an \(\alpha\) helix separated from another \(\alpha\) helix by a turn, called a helix-turn-helix unit, is found in many proteins which bind DNA.
Some polypeptides fold into multiple compact regions which may be connected via a flexible polypeptide segment, like pearls on a string. These compact globular units, called domains, range in size from \(30\) to \(400\) residues; for example, the extraccelular part of \(\text{CD}4\), a cell-surface protein on some cells of the immune system, comprises four similar domains of approximately \(100\) residues each. Different proteins may share domains despite differences in their tertiary structures.
Many proteins comprise multiple polypeptide chain in their functional states; each polypeptide in such a protein is named a subunit. Quaternary structure titles the arrangement of subunits and the nature of their interactions (typically weak interactions).
The simplest quaternary structure is a dimer consisting of two identical subunits; this organization is present in Cro, a DNA-binding protein found in the bacterial virus \(\lambda\). Quaternary structure can instead be as complex as dozens of unique polypeptides: more than one type of subunit can be present, often in variable numbers. For example, hemoglobin consists of two \(\alpha\) subunits and two \(\beta\) subunits, and the molecule is therefore titled an \(\alpha_2\beta_2\) tetramer.
Christian Anfinsen, in the 1950s, revealed the relationship between the linear sequence of a protein and its conformation via the study of ribonuclease, a polypeptide monomer comprising \(124\) residues cross-linked by four disulfide bonds. Anfinsen's plan was to destroy the three-dimensional structure of the enzyme, then to determine the conditions required to restore the tertiary structure: chaotropic agents such as urea were added to a solution of ribonuclease, and the disulfide bonds were cleaved reversibly with the sulfhydryl agent \(\beta\)-mercaptoethanol (in the presence of a large excess of such an agent, the disulfides are converted into sulfhydryls).
The resulting product was a denatured polypeptide. Anfinsen then observed that the denatured ribonuclease, freed of urea and \(\beta\)-mercaptoethanol, slowly regained enzymatic activity, spontaneously refolding into a catalytically active form, with all disulfide bonds reforming. The resulting conclusion was that the information needed to specify the catalytically active tertiary structure of ribonuclease is contained in its linear sequence; more generally, the native structure is also that which is most thermodynamically stable.
A random search of every possible conformation a protein could adopt would be abysmally slow. Cyrus Levinthal calculated that, assuming each residue of a \(100\)-residue protein can assume three unique conformations, the total number of structures would be \(5 \times 10^{47}\): assuming the conversion of one structure into another would take \(10^{-13}\) seconds, the resulting search time would total to \(1.6 \times 10^{27}\) years. Proteins realistically fold at a far faster pace than this, and therefore must follow a partly defined folding pathway consisting of intermediates between the entirely denatured protein and its native structure as to avoid a random search through each conformation.
The essence of protein folding is the tendency to retain partly correct intermediates, as they exhibit greater stability than unfolded regions. The criterion of correctness is not, however, a residue-by-residue scrutiny of conformation, but instead the total free energy of the folding intermediate. Even correctly folded proteins are only marginally stable: the free-energy difference between the folded and unfolded states of a typical \(100\)-residue protein is \(42 \text{ kJ mol}^{-1}\), with each residue contributing only \(0.42 \text{ kJ mol}^{-1}\) on average to maintain the folded state. For comparison, the amount of thermal energy at room temperature is \(2.5 \text{ kJ mol}^{-1}\). Correct intermediates can thus be lost; but, the average tendency is given by a positive feedback loop of increasingly stabilizing intermediates.
The folding of proteins can be visualized as a folding funnel: the breadth of the funnel represents all possible conformations of the unfolded protein, while the depth represents the energy difference between the folded and native protein. Any point on the surface represents a possible three-dimensional structure and its energy value.
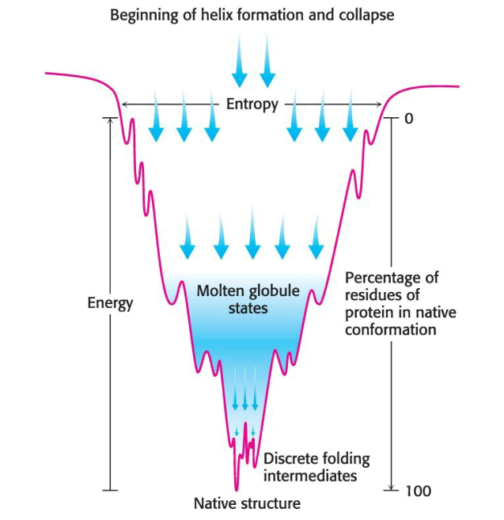
One model suggests that local interactions occur first, and it is the resulting secondary structure which facilitates the long-range interactions which lead to tertiary-structure formation. Another model suggests that the hydrophobic effect draws together hydrophobic amino acids in far proximity from one another, leading to the formation of a globular structure: as the hydrophobic interactions are presumed to by dynamic, the structure is named a molten globule. A third model, titled the nucleation-condensation model, involves both of the previous models as factors in the formation of the native state.
It has been known for some time that certain proteins can adopt two unique structures, one of which results in protein aggregation and pathological conditions; such alternate structures originating from a unique linear sequence were thought to be rare, but recent work has called into question the universality of the idea that each linear sequence gives rise to one structure.
Intrinsically disordered proteins, entirely or in part, do not have a discrete three-dimensional structure under physiological conditions. An estimated \(50 \%\) of eukaryotic proteins have at least one unstructured region greater than \(30\) residues in length. These proteins assume defined structure upon interaction with other proteins, permitting one protein to assume unique structures in its interactions with different partners. IDPs are especially crucial in signaling and regulatory pathways.
Metamorphic proteins appear to exist in an ensemble of structures of approximately equal energy, in equilibrium. Small molecules or other proteins may bind to a particular member of the ensemble, resulting in a complex with a biochemical function unique to that of another complex formed via the binding of the same Metamorphic protein to a different partner. Cytokine lymphotacin, part of a class of signal molecules in the immune system which bind to receptor proteins on the surface of immune-system cells, exists in two highly unique structures which exist in equilibrium: one structure is a characteristic of chemokines, comprising a three-stranded \(\beta\) sheet and a carboxyl-terminal helix, which binds to its receptor and activates it; the alternative structure is an identical dimer of all \(\beta\) sheets, adopting which form lymphoactin binds to glycoaminoglycan, a complex carbohydrate. The biochemical activities of either structure are unique and mutually exclusive; yet, both activities are required for full biochemical activity of cytokine.
Note that in some cases, a gene can encode a single protein with more than one structure or function, effectively expanding the protein-encoding capacity of the genome.
The first step toward understanding the utility of proteins in vivo is to study their behavior in vitro: the proteins must be separated from the irrelevant constituents of the cell so that their biochemical properties can be identified and characterized. It is several techniques which allow the linear sequence of a protein to be elucidated.
The completely sequenced human genome comprises \(3\) billion bases and about \(20000\) protein-encoding genes; this genomic knowledge is analogous to a list of parts for a car, in which the details of the mechanistic intertwining of each part is absent. The proteome represents the functional information derived from proteins expressed by the genome.
Unlike the genome, the proteome is not a fixed characteristic of the cell: it represents the functional expression of information, and thus varies with cell type, developmental stage, and environmental conditions. The proteome may be mutated also by the enzymatic modifications to proteins and the interactions between proteins to form functional complexes.
A protein of interest must be isolated from the thousands of irrelevant proteins present in the cell to allow examination and characterization. The resulting sample may be only a fraction of \(1\%\) of the starting material, whether that starting material consists of cells in culture or a particular organ.
An assay checks the success of each protein purification step; for enzymes, the assay is typically based on the reaction catalyzed by the enzyme in the cell.
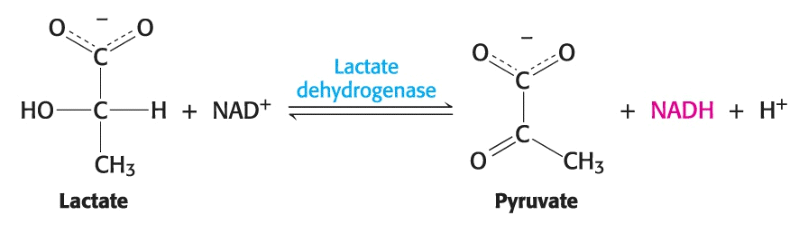
In the above reaction, lactase dehydrogenase catalyzes the transformation of lactase to reduced nicotinamide adenine dinucleotide (NADH), which, uniquely among the reaction components, absorbs light at \(340 \text{ nm}\). We can follow the process of the reaction by measuring light absorbance at \(340 \text{ nm}\), for instance, within one minute after the sample that contains the enzyme; this is our assay for the purification of lactate dehydrogenase. Note that the assay offers the magnitude of enzyme activity rather than that of enzyme protein.
To be certain of the success of the purification scheme, we require an additional piece of informaton: the amount of total protein present in the mixture being assayed. We can access the progress of our purification by measuring the specific activity --- the ratio of enzyme activity to the amount of protein enzyme in the enzyme assay --- at each step of our purification. The objective of purification to remove all irrelevant proteins expresses quantitatively the maximization of specific activity.
Once an assay is found, the cells must be broken open to release their cellular contents for analysis. The disruption of cell membranes yields a homogenate --- a mixture of the components of the cell, but no intact cells --- centrifuged at low centrifugal force, yielding a pellet of heavy material at the bottom of the centrifuge tube and a lighter solution above, coined the supernatant.
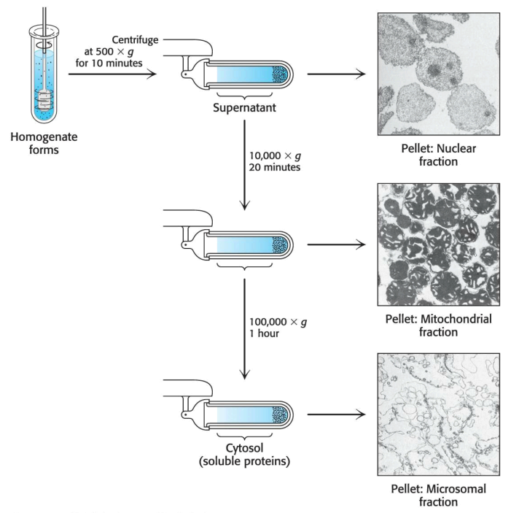
The pellets are enriched in a particular organelle. The pellet and supernatant are referred to as fractions, as we are fractionating the homogenate. The supernatant is centrifuged again at a greater force to yield yet another pellet and supernatant. This process --- differential centrifugation --- yields several fractions of decreasing density, each containing hundreds of different proteins which can be assayed for the activity being purified. Frequently, one fraction --- the crude extract --- will have the greatest enzyme activity; it then serves as the source of material to which more-discriminating purificaton techniques are applied.
Protein mixtures are typically subjected to a series of separations, each based on a different property.
Most proteins require salt to dissolve, a process coined salting in; but, they instead precipitate out of solution at high salt concentration, an effect termed salting out.
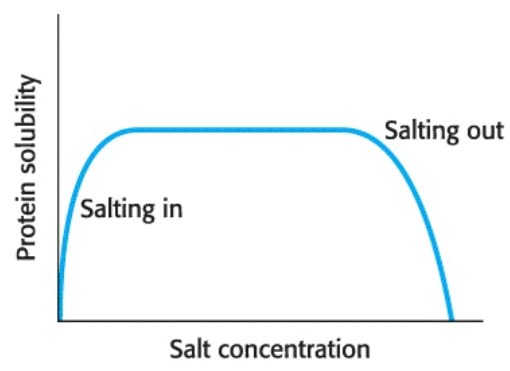
Salting out occurs due to competition between the salt ions and the protein for water to keep the protein in solution --- the water of solvation. The salt concentration at which a protein precipitates differs from one protein to another; hence, salting out can be used to fractionate a mixture of proteins.
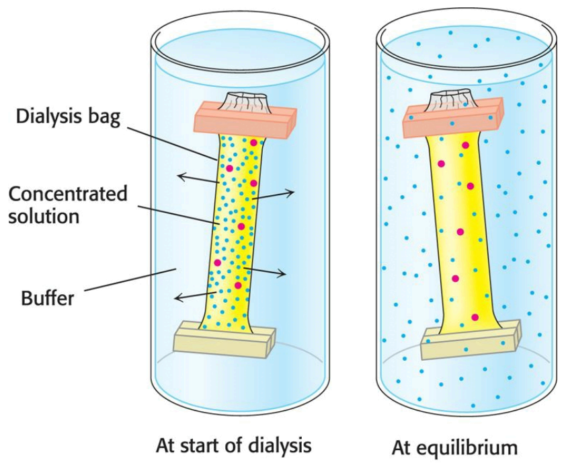
Many proteins lose their activity in the presence of such high concentrations of salt; fortunately the salt can be removed via dialysis. The protein-salt solution is placed in a small bag made of a porous, semipermeable membrane (such as a cellulose membrane). Proteins cannot fit through the pores of the membrane, whereas smaller molecules and ions such as salts can escape to emerge in the medium outside the bag (the dialysate).
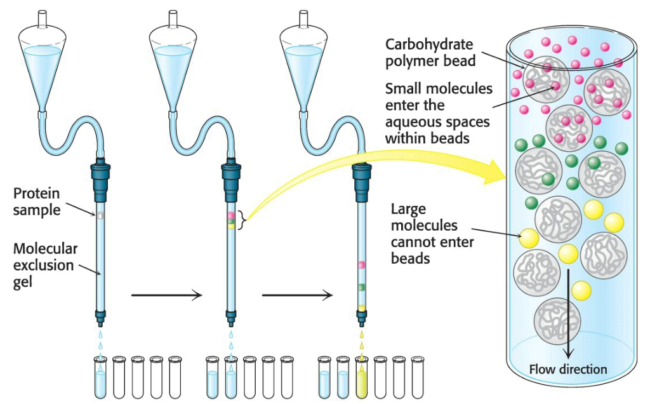
Molecular exclusion chromatography (or gel-filtration chromatography) separates proteins via size. The sample is applied to the top of a column consisting of porous beads comprising an insoluble polymer such as dextran, agarose, or polyacrylamide. Small molecules can enter these beads while large ones cannot; those larger molecules follow a shorter path to the bottom of the column and thus emerge first. Molecules which are of a size to occasionally enter a bead will flow from the column at an intermediate position, while small molecules, which take a longer, more circuitous path, will exit last.
Proteins can be separated via their net charge; if a protein has a net positive charge at \(\text{pH } 7\), it will usually bind to a column of beads containing negatively charged carboxylate groups, whereas a negatively charged protein will not bind to the column.
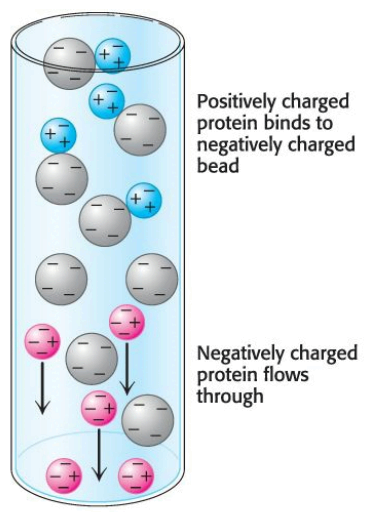
A positively charged protein bond to such a column can then be released by increasing the concentration of salt in the buffer poured over the column. The cations of the salt compete with positively charged groups on the protein for binding to the column; likewise, a protein with a net negative charge will be bound to ion-exchange beads carrying positive charges and can be eluyed from the column with the use of a buffer containing salt.
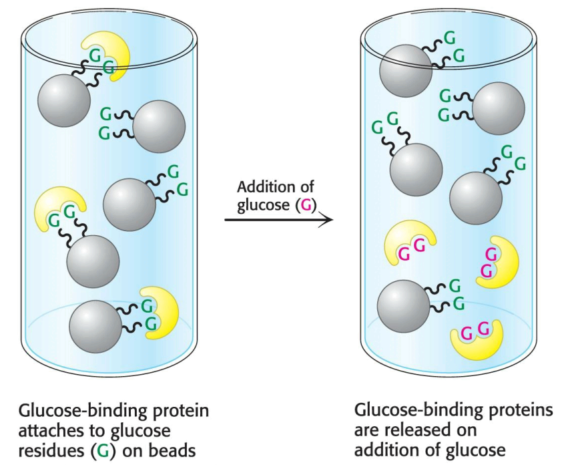
Some proteins have a high affinity for specific chemical groups or molecules; for example, the plant protein concanavalin A, which binds to glucose reversibly, can be purified by passing a crude extract through a column of beads containing covalently attached glucose residues. Concanavalin A binds to such beads, whereas most other proteins do not. The bound concanavalin A can then be released from the column by adding a concentrated solution of glucose. The glucose in solution displaces the column-attached glucose residues from binding sites on concanavalin A.
The ability of column techniques to separate individual proteins --- the resolving power --- can be improved substantially via a technique coined high-pressure liquid chromatography (HPLC), an enhanced version of the column techniques already discussed. The beads that make up the column material themselves are much more finely divided; as a consequence, there are more interaction sites and thus greater resolving power. As the column is made of finer material, pressure must be applied to the column to obtain adequate flow rates. The net result is high resolution as well as rapid separation.
We can tell a purification scheme is effective via the demonstration that the specific activity rises with each purification step, or via the visualization of the number of proteins present at each step. Gel electrophoresis permits the latter method.
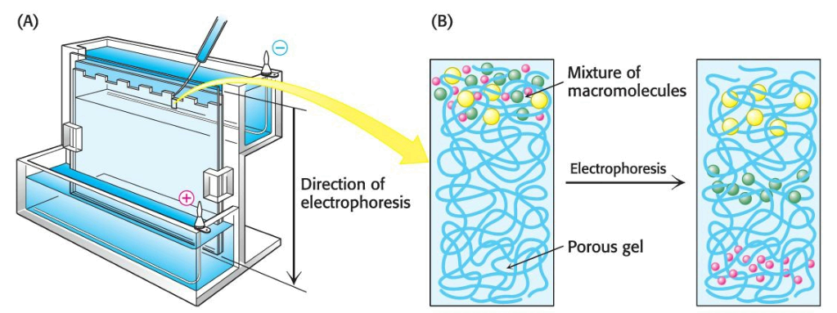
A molecule with a net charge will move in an electric field, a phenomenon termed electrophoresis. The distance and speed that a protein moves in electrophoresis depends on the electric-field strength, the net charge on the protein --- which is a function of the \(\text{pH}\) of the electrophoretic solution --- and the shape of the protein. Electrophoretic separations are nearly always carried out in gels such as polyacrylamide; the gel serves as a molecular sieve which enhances separation via size. The electrophoresis of proteins is performed in a thin, vertical slab of polyacrylamide. The \(\text{pH}\) of the electrophoretic solution is adjusted so that all proteins are negatively charged. The direction of flow points from the cathode (negative charge) to the anode (positive charge).
Proteins can be separated largely via mass by electrophoresis in a polyacrylamide gel in the presence of the detergent sodium dodecyl sulfate (SDS), a technique called SDS-polyacrylamide-gel electrophoresis (SDS-PAGE). The anionic SDS denatures proteins and binds to the denatured protein at a constant ratio of one SDS molecule for every two amino acids in the protein. The negative charges on the many SDS molecules bound to the protein swamp the normal charge on the protein, giving all proteins the same charge-to-mass ratio; thus, proteins will differ only in their mass. Finally, a sulfhydryl agent such as mercaptoethanol is added to reduce disulfide bonds and entirely linearlize the proteins. The SDS-protein complexes are then subjected to electrophoresis; when this is complete, the proteins in the gel can be visualized by staining them with silver or a dye such as Coomassie blue, which reveals a series of bands. Small proteins move rapidly through the gel, whereas large proteins stay at the top, near the point of application of the mixture.
Proteins can also be separated electrophoretically via the relative contents of their acid and basic residues. The isoelectric point (\(\text{pI}\)) of a protein is the \(\text{pH}\) at which its net charge is zero; at this \(\text{pH}\), the protein will not migrate in an electric field.
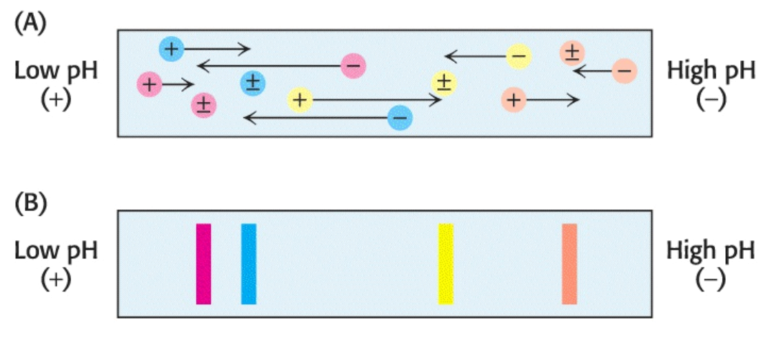
If a mixture of proteins is subjected to electrophoresis in a \(\text{pH}\) gradient in a gel in the absence of SDS, each protein will move until it reaches a position in the gel at which \(\text{pH}\) is equal to the \(\text{pI}\) of the respective protein. This method of separating proteins is called isoelectric focusing. Proteins differing by one net charge can be separated.
Isoelectric focusing can be combined with SDS-PAGE to obtain very high resolution separations. A single sample is first subjected to isoelectric focusing; this single-lane gel is then placed horizontally on top of an SDS-polyacrylamide slab, then subjected to electrophoresis again, in a direction perpendicular to the isoelectric focusing, to yield a two-dimensional pattern of spots.
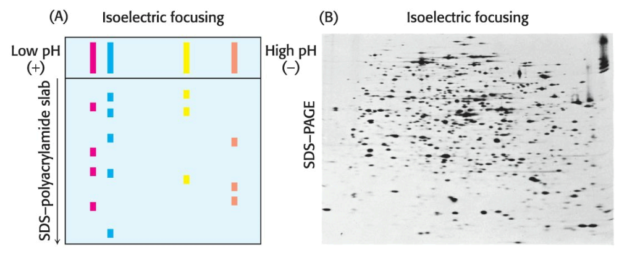
In such a gel, proteins have been separated in the horizontal direction via \(\text{pI}\) and in the vertical direction via mass. Over a thousand unique proteins in the bacterium Escherichia coli can be resolved in a single experiment via two-dimensional electrophoresis.
Proteins isolated from cells under different physiological conditions can be subjected to two-dimensional electrophoresis. The intensities of individual spots on the gels can then be compared, which indicates that the concentrationsof specific proteins have changed with physiological state. It is thus possible to identify proteins by coupling two-dimensional gel electrophoresis with mass spectrometry, a highly sensitive technique for the determination of the precise mass of the proteins in a given sample.
Some combination of purification techniques will usually yield a pure protein; to determine the success of a protein-purification, we monitor the procedure at each step by determining specific activity and by performing an SDS-PAGE analysis.
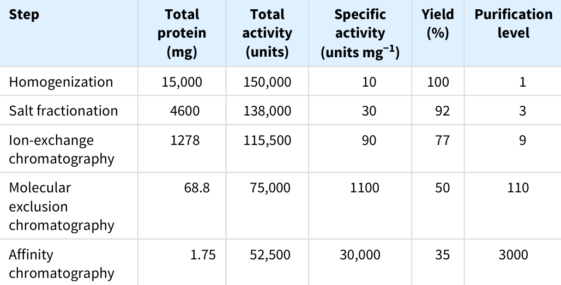
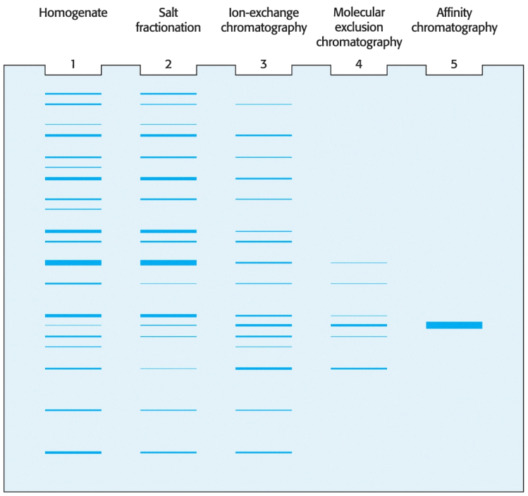
Consider the results for the purification of the above hypothetical protein. At each step, the following parameters are measured:
Inevitably, some of the protein is lost in each purification step, such that our total yield is \(35\%\). A good purification scheme takes into consideration purification levels as well as yield.
Notably, the SDS-PAGE depicted above shows that if we load the same amount ofprotein onto each lane after each step, the number of bands decreases proportionally to the level of purification, and the amount of protein of interest increases as a proportion of the total protein present.
The estrogen-receptor protein binds the the female steroid hormone estradiol, an estrogen, then regulates the expressions of genes which play a role in the development of the female phenotype. But, the estrogen receptor has no enzyme activity.
The estrogen receptor is the only protein in estrogen-responsive tissues which can bind to the estradiol with high affinity; we can exploit this distinctive property by exposing the cytosol to radiolabeled estradiol. Because the estrogen receptor has such a great affinity for estradiol, it will be unique among proteins in the cell in its binding to this radioactive steroid. Measurement of this phenomenon requires a second part to this assay: a means to detect the estradiol-receptor complex.
Ultracentrifugation permits the separation of very small molecular complexes. Proteins or protein complexes will move in a liquid medium when subjected to a centrifugal force: the rate at which these complexes or particles move when exposed to such force is determined by their respective mass, density, and shape.
We can calculate the sedimentation coefficient of a particle to quantify its rate of movement given
\[s = \frac{m(1 - \bar{v}\rho)}{f}\]
where:
The \(1 - \bar{v}\rho\) term is the buoyant force exerted by the liquid medium. Sedimentation coefficients are usually expressed in Svedberg units (\(\text{S}\)), equal to \(10^{-13}s\). The magnitude of \(\text{S}\) correlates with the speed of the molecule through a centrifugal field. Several important conclusions can be drawn from the preceding equation:
We can begin characterizing a protein's movement through a centrifugal field via the formation of a density gradient in a centrifuge tube. Differing proportions of a low- and high-density solution --- such as \(5\%\) and \(20\%\) sucrose, respectively --- are mixed to create a linear gradient of sucrose concentration ranging from \(20\%\) at the bottom of the tube to \(5\%\) at the top. The role of the gradient is twofold: first, the gradient stabilizes the liquid and prohibits movement of the liquid due to convection; second, the resistance to movement resulting from the increasing density of the gradient helps to improve separation of the sample components.
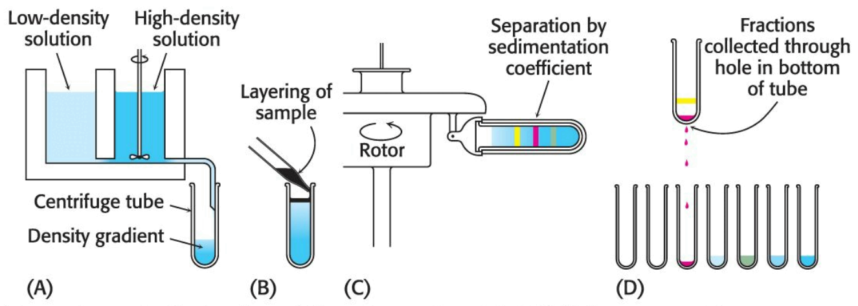
Next, a small volume of solution containing the mixture of proteins to be separated --- which includes the radiolabeled estradiol-receptor complex --- is placed on top of the density gradient. When the rotor is spun proteins move through the gradient and separate based on their sedimentation coefficients. The time and speed of the centrifugation is determined empircally.The separated bands (or zones) of protein can be harvested by making a hole in the bottom of the tube and collecting the solution via drops, which can each be measured for their respective protein content, catalytic activity, or another functional property. In regard to the estrogen receptor, the fractions are measured for radioactivity.
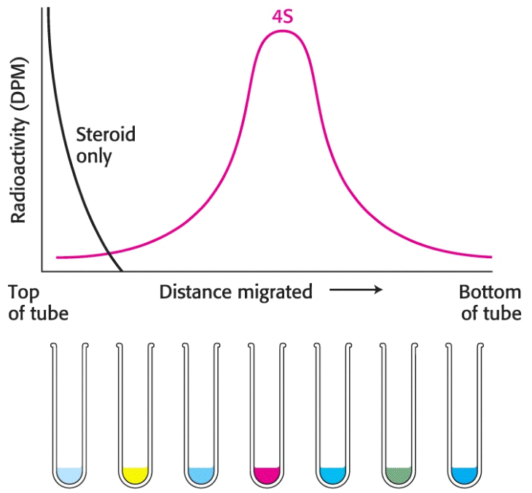
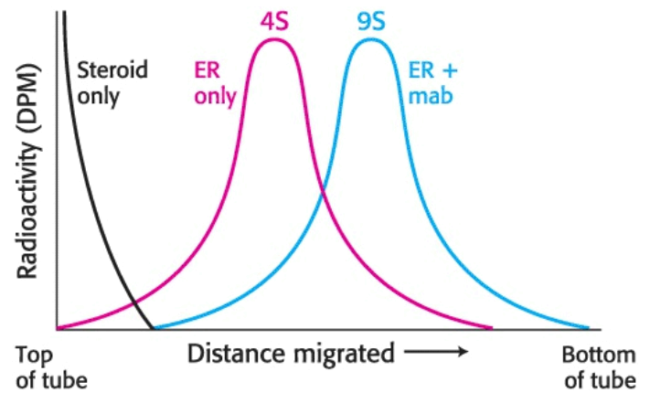
Radiolabeled estradiol alone is far too small to move under centrifugal force: the radioactivity must be associated with the receptor. A comparison with standards established that the \(\text{S}\) value of the receptor was \(4\); however, hundreds of proteins have an \(\text{S}\) value near \(4\). Thus, although the radioactivity profile shows only the receptor, there are indeed many other proteins in the same region of the centrifuge tube; although we can only identify the receptor, it regardless is an impure collection of proteins.
Now, with an assay for the receptor, we must determine a strategy for purifying the receptor: as this protein proved difficult to purify with the use of the methods previously described, we will use immunological techniques.
Immunological techniques begin with the generation of antibodies to a particular protein. An antibody (or immunoglobulin) is itself a protein; it is synthesized by an animal in response to the presence of a foreign substance, called an antigen.
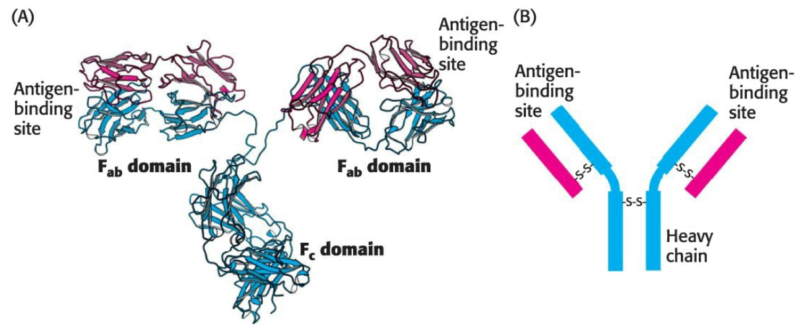
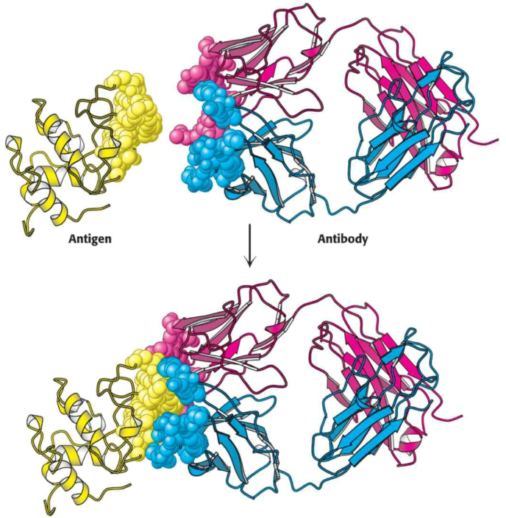
Antibodies have specific and high affinity for the antigens which elicted their synthesis. The binding of antibody and antigen is a step in the immune response which protects the animal from infection.
Proteins, polysaccharides, and nucleic acids can be effective antigens. When a protein is the antigen, an antibody recognizes the specific group or cluster of amio acids on the target molecule, termed an antigenic determinant (or epitope). Animals have a great repertoire of antibody-producing cells, each synthesizing an antibody of a single specificity. The binding of antigen to antibody simulates the proliferation of the small number of cells which had already been forming an antibody capable of recognizing the antigen.
Immunological techniques depend on the ability to generate antibodies to a specific antigen. To obtain antibodies which recognize a particular protein, we can inject the protein into a test subject --- a rabbit, for example. The injected protein stimulates the reproduction of cells producing antibodies that recognize it. Blood is drawn from the immunized rabbit several weeks later and centrifuged to separate blood cells from the supernatant, coined the serum (blood from which the cells have been removed); the serum, termed an antiserum, contains antibodies to all antigens to which the rabbit has been exposed. Only some antibodies will be for the injected protein; moreover, frequently, many unique antibodies can bind to a single antigen.
Animals produce many unique, heterogenous (or polyclonal) antibodies, each recognizing a respectively unique surface feature of the same antigen. The heterogeneity of polyclonal antibodies can be advantageous in applications such as the detection of a protein of low abundance, as each protein molecule can be bound by more than one antibody at multiple distinct antigenic sites.
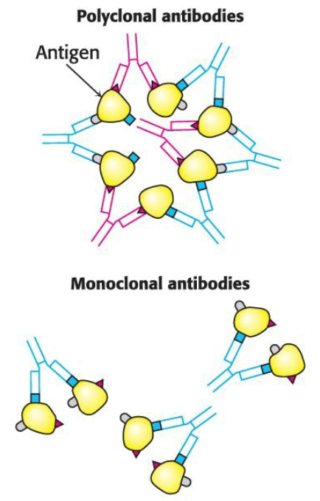
Just as working with impure proteins, working with an impure mixture of antibodies impedes characterization; rather than separate the target antibody from the mixture --- as we would with a protein --- we will find it more effective to isolate a group of identical cells which produce a single kind of antibody. A barrier is that antibody-producing cells die quickly in vitro, leaving little product with which to work.
Immortal cell lines which produce monoclonal antibodies do exist, and are derived from a type of cancer: multiple myeloma, a malignant disorder of antibody-producing cells. In this cancer, a single transformed plasma cell divides uncontrollably, generating a great number of cells of a single kind. Such a group of cells is a clone, as the cells are descended from the same cell and thus share all properties. Each generation of the identical cells of the myeloma secrete large amounts of normal antibodies of a single kind; however, although these antibodies were useful for elucidating antibody structure, nothing is known about their specificity, prohibiting their use in immunological methods.
Cesar Milstein and Georges Kohler discovered that large amounts of a homogenous antibody of nearly any desired specificity can be obtained by fusing a short-lived antibody-producing cell with an immortal myeloma cell. For example, an antigen is injected into a mouse, and its antibody-producing spleen is removed several weeks later. A mixture of plasma cells from the spleen is fused in vitro with myeloma cells; each of the resulting hybrid cells, coined hybridoma cells, indefinitely produces the identical antibody specified by the parent cell from the spleen. Hybridoma cells will be produced for each protein which was injected into the mouse. The cells can then be screened, using an assay specific to he antigen-antibody interaction, to determine which ones produce antibodies having hte desired specificity. This process is repeated intil a pure cell line --- a clone producing a single antibody --- is isolated.
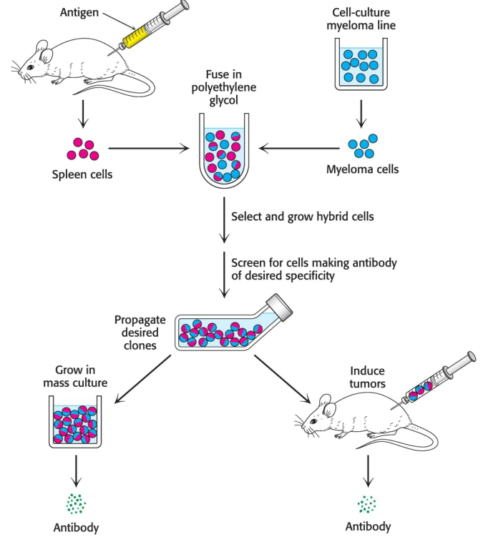
The next objective is to generate a monoclonal antibody to the estrogen receptor, then to use the antibody to isolate the receptor; we inject a small amount of cytosol from the rat uterus to generate hybridoma cells as just described. We now have a population of antibodies, some of which are specific for the estrogen receptor.
We can detect estrogen-receptor-specific antibodies via gradient centrifugation: if we mix the antibody preparation with the cytosol containing the radiolabeled estradiol-receptor complex, an antibody will bind to the estrogen receptor. This binding will alter the sedimentation profile obtained when we centrifuged the radiolabeled estradiol-receptor complex.
When a estradiol-receptor complex is incubated with antibodies, the complex of radioactive steroid and estrogen receptor undergoes sedimentation at \(9\text{S}\) in a sucrose gradient, in association with the antibody.
The population of antibody-producing cells is screened for those that are generating antibodies which bind the estradiol-receptor complex. After a certain number of screens, we will have the pure line --- a monoclonal cell line --- which is producing only one antibody, coined a monoclonal antibody, to the estrogen receptor.
We can isolate pure estrogen receptors from the morass of proteins in the cell cytosol by fishing for them, using monoclonal antibodies as bait. The monoclonal antibody can be covalently linked to insoluble beads